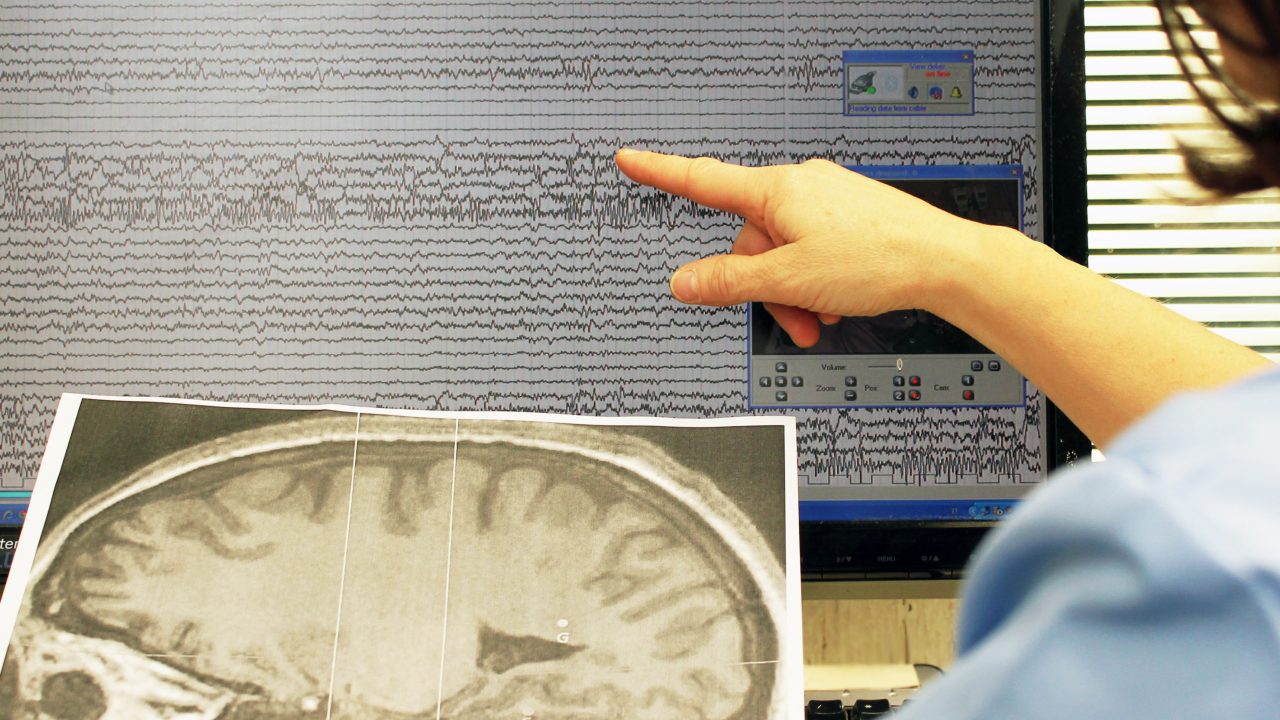
Medicina & Salute Malattie neurodegenerative: un nuovo farmaco blocca la progressione della CLN2 Views 25
{kind=link}